Тетрада Фалло
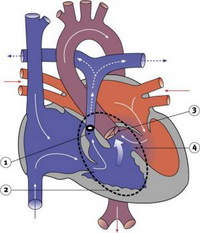 Тетрада Фалло — это врожденный порок сердца, классически имеет 4 анатомических компонента: дефект межжелудочковой перегородки, обструкцию выводного тракта правого желудочка, дестрапозицию аорты и гипертрофию миокарда правого желудочка.
Тетрада Фалло — это врожденный порок сердца, классически имеет 4 анатомических компонента: дефект межжелудочковой перегородки, обструкцию выводного тракта правого желудочка, дестрапозицию аорты и гипертрофию миокарда правого желудочка.
Первые сообщения о таком сочетании пороков можно найти в работах анатома Стенсена в 1671 году. В 1888 году врач Фалло описал сочетание этих аномалий в одном сердце как отдельную болезнь. Позже болезнь была названа в его честь.
Встречается Тетрада Фалло в 6,5-9,7% случаев от всех врожденных пороков сердца. Клинически до месячного возраста порок проявляется у 65% детей — это один из »синих» пороков. Название говорит само за себя. При »синих» пороках дети или периодически синеют, или имеют такой цвет всегда. Этот симптом является следствием того, что по каким-то причинам, в большом круге кровообращения циркулирует бедная кислородом кровь и ребенок страдает от кислородного голодния (гипоксии). В норме венозная кровь попадает в правые отделы сердца и выбрасывается в легочную артерию для обогащения кислородом в легких. При этом пороке венозная кровь не способна вся пройти через узкую легочную артерию в легкие. Часть венозной крови через дефект межжелудочковой перегородки попадает непосредственно в левый желудочек, а из него в аорту. Чем меньше диаметр легочной артерии или выхода из правого желудочка тем больше венозной крови подмешивается к артериальной и разносится ко всем органам и тканям. Поэтому дети выглядят синими, а в тяжелых случаях у них возникают одышечно-цианотические приступы, которые могут привести к потере сознания и смерти ребенка. К счастью, этот порок давно и успешно лечится хирургическим путем.
Первая операция выполнена в 1945 году хирургом Блэлоком с США. Идею этой операции хирургу подсказала педиатр Элен Тауссиг. Операция до сих пор актуальна и названа в честь своих изобретателей — Блэлок-Тауссиг анастомоз. Эта операция не радикальная, не исправляет порок, но помогает ребенку дожить без гипоксии до следующего этапа — радикальной коррекции. При неосложненной форме Тетрады Фалло хирургическое лечение возможно в один этап. В условиях искусственного кровообращения сразу выполняется радикальная коррекция порока.
В тяжелых случаях без медицинской помощи на первом году жизни умирает до 25% детей, затем еще 40% умирает до 3 лет, 70% — до 10 лет. Однако, риск хирургического лечения составляет лишь 3-6%.
Коарктация аорты
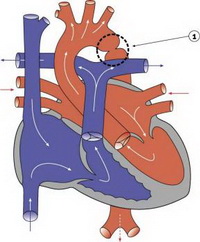 Коарктация аорты – это врожденное сужение аорты, чаще всего в области артериального протока. Порок встречается как в изолированном виде так и в сочетании с другими пороками сердца.
Коарктация аорты – это врожденное сужение аорты, чаще всего в области артериального протока. Порок встречается как в изолированном виде так и в сочетании с другими пороками сердца.
Порок впервые описал анатом Мекель в 1750 году, название аномалии «коарктация» предложили почти через столетие. Первая успешная операция была проведена в 12-летнего мальчика шведским хирургом Крафордом еще в 1944 году.
Коарктация аорты встречается у 8% детей с пороками сердца. Почти в 3 раза чаще болеют мальчики чем девочки. Уже во время внутриутробного развития коарктация аорты вызывает патологические изменения в миокарде плода.
Специфической является гемодинамика этого порока. От аорты к верхней половине туловища отходят 3 сосуда, за ними находится сужение, которое может быть резким или даже критическим. Для того, чтобы преодолеть такую преграду, сердцу нужно развивать большую силу при каждом сокращении. Поэтому в аорте и ее сосудах выше места сужения возникает высокое давление. У новорожденного ребенка артериальное давление на руках может достигать 200 мм рт ст, при том, что нормальное давление на руках у здорового младенца около 50 мм рт ст. Однако ниже места сужения в аорте низкое давление, на ногах пуль ослаблен или отсутствует. Все органы и ткани нижней половины туловища и ноги развиты недостаточно. Если не прооперировать такого ребенка, то со временем, в зависимости от критичности сужения, резко снижается сократительная способность миокарда, вплоть до остановки сердечной деятельности. При коарктации аорты смертность детей на первом году жизни без медицинской помощи может достигать 75%.
Помощь таким детям одна — срочная операция. Цель хирургического лечения — полное восстановление проходимости аорты. Операция выполняется без аппарата искусственного кровообращения, на работающем сердце. Выше и ниже сужения аорта полностью пережимается зажимами, удаляется узкое место, и два конца аорты между собой сшиваются. После снятия зажимов ток крови по аорте восстанавливается. Риск оперативного вмешательства около 1,5-2%.
Дефект межжелудочковой перегородки
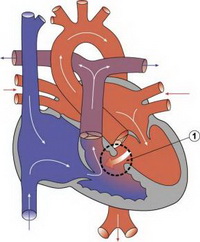 Дефект межжелудочковой перегородки – это врожденный порок сердца, при котором есть сообщение между правыми и левыми камерами сердца на уровне желудочков.
Дефект межжелудочковой перегородки – это врожденный порок сердца, при котором есть сообщение между правыми и левыми камерами сердца на уровне желудочков.
Первые описания анатомии порока относятся к 1872 году. Первая успешная операция по закрытию межжелудочкового дефекта выполнена Лилехаем в 1995 в США. Этот дефект может существовать как самостоятельный порок, так и быть компонентом более сложной сердечной аномалии (Тетрада Фалло, транспозиция магистральных сосудов, коарктация аорты и др).
Дефект межжелудочковой перегородки — это самый часто встречаемый порок сердца. По данным различных авторов его обнаруживают у 15-25% детей с пороками сердца, или у трех детей на 2000 всех родившихся детей.
Нарушение гемодинамики при дефекте межжелудочковой перегородки зависит от его размера или количества дефектов. В норме давление в правом желудочке во время сокращения в 3-4 раз ниже, чем в левом, поэтому происходит сброс крови слева направо при каждом сокращении сердца. Если дефект межжелудочковой перегородки большой (от 1см) клинические проявления видны с рождения.
У маленького ребенка наблюдается постоянная одышка (даже во сне), он устаёт во время еды, у него увеличивается печень, он отстает в росте и не набирает вес. Без лечения такой ребенок может погибнуть в первые месяцы жизни. При маленьких и средних дефектах клинические проявления наступают позже и они не такие выраженные. Некоторые дефекты, расположенные в мышечной части перегородки, могут самостоятельно закрыться до 3-4 лет. Все дефекты, которые не закроются самстоятельно или имеют размеры более 3мм обязательно должны лечиться хирургическим путем. При очень большом дефекте, когда имеются лишь остатки межжелудочковой перегородки, образуется общий желудочек сердца.
Дефект межжелудочковой перегородки закрывают специальной заплатой в условиях искусственного кровообращения при полной остановке сердечной и легочной деятельности. Если дефектов много или перегородки практически нет и закрыть такие дефекты заплатой не представляется возможным, тогда выполняется другая операция без искусственного кровообращения. Специальной тесьмой производится суживание легочной артерии, что дает возможность защитить сосуды легких от высокого давления и предохранить от склерозирования.
Риск оперативного лечения зависит от возраста ребенка, размера дефекта и состояния легких и выбора операции и, как правило, не превышает 3%.
Дефект межпредсердной перегородки
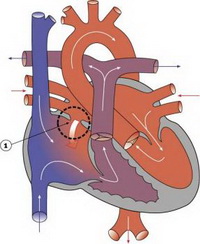 Дефект межпредсердной перегородки — врожденный порок сердца, при котором имеется сообщение между левым и правым предсердием. При наличии такого сообщения при каждом сокращении сердца происходит сброс крови из левого предсердия в правое.
Дефект межпредсердной перегородки — врожденный порок сердца, при котором имеется сообщение между левым и правым предсердием. При наличии такого сообщения при каждом сокращении сердца происходит сброс крови из левого предсердия в правое.
Первое описание аномалии принадлежит Леонардо Да Винчи в 1513 году, а более детальное описание встречается в 1826 году в работах анатома Льюиса. Первая операция выполнена в 1948 г. без искусственного кровообращения, а в 1953 г. — с использованием аппарата искусственного кровообращения.
Среди врожденных пороков сердца этот дефект встречается в 8% случаев.
Основным признаком нарушения гемодинамики при дефекте межпредсердной перегородки является сброс артериальной крови из левого предсердия в правое. Объем сброса крови и наличие клинических симптомов зависит от размера или количества дефектов. Длительное поступление большого количества крови в правые отделы сердца и сосуды легких приводят к тяжелым осложнениям в виде перегрузки правых отделов сердца и повышенного давления в легочной артерии. Это в свою очередь приводит к таким осложнениям, как сердечная слабость и развитие легочной гипертензии.
При неосложненном течении болезни больные жалуются на одышку, сердцебиение, быструю утомляемость и склонность к простудным заболеваниям. Первые проявления болезни чаще возникают в 2-3 года.
Единственным эффективным методом лечения является закрытие дефектов различными методами. Наиболее распространенным методом является закрытие дефекта во время кардиохирургической операции с подключением аппарата искусственного кровообращения. С помощью современных технологий в последние годы стало возможным лечение этой аномалии без разреза (ендоваскурярним методом). Через сосуды бедра специальное устройство проводят к сердцу и закрывают им дефект под контролем рентгена.
Средняя продолжительность жизни пациентов с большими дефектами межпредсердной перегородки не превышает 27 -30 лет, со средними — 36-40 лет. После закрытия дефекта 80% паиентов становятся вполне здоровыми людьми. Риск радикальной коррекции не превышает 0,5%.